|
|
Home > Problems > Chapter 7
| The programs normally used for phylogenetic prediction are the PHYLIP programs or PAUP. For simple data sets, the tree may be calculated by hand as illustrated below. |
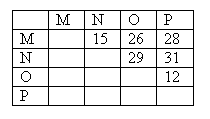
1. The following distances are found among four sequences M, N, O, and P:
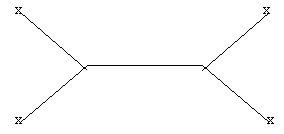
- Draw all possible unrooted trees for the four sequences M, N, O, and P by labeling the X's.
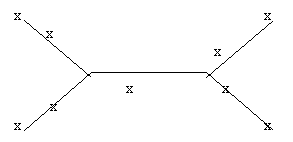
- Based on the most closely related sequences in the table, choose the most likely tree.
- Label the tree branches with lowercase letters m, n, o, p, and q (where the lowercase x is indicated), being the branch going to node M, etc.:
- Calculate the lengths of the tree branches using the Fitch�Margoliash method described in the class notes.
- Calculate the branch lengths and the position of the tree root using the UPGMA method as described in the class notes.
- What assumptions are made by the above methods regarding mutation rates in the different branches of the evolutionary tree?
|
|
|
| © 2004 by Cold Spring Harbor Laboratory Press. All rights reserved. |
 |
| No part of these pages, either text or image, may be used for any purpose other than personal use. Therefore, reproduction, modification, storage in a retrieval system, or retransmission, in any form or by any means, electronic, mechanical, or otherwise, for reasons other than personal use, is strictly prohibited without prior written permission. |
|
|
|
